A groundbreaking new model known as GraphBAN offers promising advancements in the field of drug discovery by significantly improving the ability to predict compound-protein interactions (CPIs). In a detailed study led by Hadipour et al., published in Nature Communications, researchers explored the intricate relationships between various compounds and target proteins, ultimately providing a powerful tool to assist in identifying potential therapeutic agents.
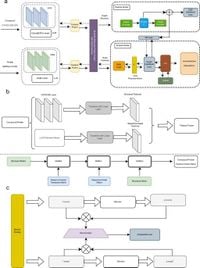
The GraphBAN framework, distinctively a graph-based approach, incorporates a knowledge distillation architecture, which features a teacher-student model designed to predict interactions inductively. This innovative model adeptly handles inductive link predictions between entirely unseen compounds and proteins, marking a significant evolution from traditional methodologies that often rely on known contexts.
Drug discovery is inherently resource-intensive and fraught with challenges surrounding the identification of suitable drug candidates. Computational methods have typically aimed to alleviate these burdens, yet many existing models struggle with the complexities of diverse datasets or the identification of novel interactions amongst unseen compounds and protein targets. GraphBAN addresses these challenges head-on, employing a domain adaptation module to enhance its performance across different dataset domains.
According to the authors of the article, “Empirical tests on five benchmark datasets demonstrate that GraphBAN outperforms ten baseline models.” This claim is supported by extensive evaluations where GraphBAN showcased its effectiveness in predicting CPIs across various datasets such as BindingDB, BioSNAP, C.elegans, KIBA, and PDBbind 2016.
To delve into its workings, GraphBAN uses a unique methodology that blends structural graph convolutional networks (GCN) and a state-of-the-art pre-trained language model known as ChemBERTa to extract compound-specific features. Simultaneously, it integrates a convolutional network along with a language model for proteins, designated as evolutionary scale modeling (ESM). This dual-faceted extraction process allows it to leverage both chemical and biological insights while maintaining high predictive performance.
In practical applications, GraphBAN underwent rigorous testing. Its case study involved interactions with the peptidyl-prolyl cis-trans isomerase NIMA-interacting 1 (Pin1), which plays a pivotal role in cellular processes and is a promising target for cancer therapies. Utilizing a dataset of approximately 250,000 compounds from ZINC-250K, GraphBAN successfully identified numerous compounds exhibiting significant interaction probabilities with Pin1.
The study resulted in the identification of 134 promising compounds with interaction probabilities above 0.5. After rigorous filtering based on criteria such as drug-likeness properties, a final selection of nine candidates emerged, suggesting their potential use as novel therapeutics. Notably, the model “effectively captures the binding site regions,” indicating its practical utility in real-world drug discovery scenarios.
Overall, the introduction of GraphBAN presents a substantial contribution to the methodology of drug discovery, highlighting the dichotomy between traditional experimental approaches and computational efficiencies. The integration of advanced machine learning techniques, particularly knowledge distillation and domain adaptation, equips GraphBAN with the tools necessary for tailoring predictions to accurately reflect the dynamic nature of compound-protein interactions.
Looking ahead, continuous enhancements to GraphBAN's architecture and broader dataset incorporation could yield even more reliable results, integrating insights from genomic and transcriptomic data. Such expansions may refine its predictive capabilities further, cementing its role in personalized medicine and innovative therapeutic development.
As encapsulated by the researchers in their conclusion, “Our proposed GraphBAN exhibits notable performance by providing acceptable CPI prediction accuracy,” underscoring the model's significance in revolutionizing the landscape of computational drug discovery.