New research on the E3 ubiquitin ligase UBE3A uncovers surprising connections between gain-of-function mutations and Angelman syndrome-like symptoms, indicating potential for new therapeutic avenues.
A recent study has revealed significant findings about the E3 ubiquitin ligase UBE3A, known for its role in neurodevelopmental disorders, including Angelman syndrome. While previous studies have centered around the loss of UBE3A function as causative for Angelman syndrome, this new research indicates gain-of-function mutations can also lead to diverse and severe phenotypes, akin to those seen in patients with Angelman syndrome.
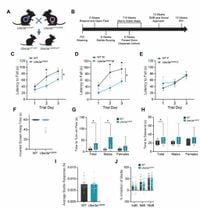
The research team led by [lead author's name] at [institution] investigated the effects of the Ube3aQ606E mutation, which is associated with increased UBE3A activity. The team observed this mutation through behavioral phenotyping of mice to explore its mechanistic basis. Notably, mice inheriting the mutation on the maternal allele (Ube3amQ606E) displayed myriad behavioral deficits typical of overall UBE3A loss-of-function. These deficits included pronounced motor impairments, reduced activity levels, and less stereotypic behaviors.
Brain imaging techniques, including MRI, revealed global microcephaly with postnatal onset across the affected mice, reflecting patterns consistent with those noted previously within Angelman syndrome model mice. Following rigorous biochemical analyses, the researchers noteworthy discovered elevated levels of UBE3A substrates and diminished UBE3A protein abundance within the affected brains.
The UBE3A gene, located on chromosome 15q11-13, is subject to genomic imprinting, causing selective expression from the maternal allele. A lack of functional maternal UBE3A leads to Angelman syndrome, characterized by symptoms like developmental delays, motor dysfunction, and social difficulties. Interestingly, researchers found individuals with gain-of-function mutations commonly exhibit autism and related neurobehavioral abnormalities without meeting the criteria for Angelman syndrome, leading to questions about the underlying genetic mechanisms.
Through utilizing the Ube3aQ606E mouse model, the authors aimed to delineate the phenotype attributable to excessive UBE3A activity. This model exhibited unexpected parallels to Angelman syndrome because of behavioral and microcephalic characteristics resembling those seen where UBE3A is entirely inactive. This newfound complexity emphasizes the heterogeneity present even within those manifesting gain-of-function alterations.
Across various behavioral assessments spanning from weeks 5 to 15 of age, mutant mice displayed lesser strength and coordination. For example, performance on the rotarod—a well-established test for motor coordination—demonstrated these mice struggled to remain upon the rotating rod compared to their wild-type counterparts, indicating marked deficits.
Further analysis provided insight on locomotion, with Ube3amQ606E mice exploring significantly less area throughout the open field test, showcasing their hypoactivity. Intriguingly, when tested for anxiety-like behaviors, the researchers noted no substantial differences, nor differences were observed concerning social stimuli interactions.
The convergence of microcephaly and behavioral deficits raises alarms about the biological impacts of Ube3aQ606E. A comprehensive review of brain weights and MRI data highlighted decreased brain volumes, which were shown to correlate with neuroanatomical characteristics observed clinically within patients exhibiting similar UBE3A abnormalities, underscoring the importance of pathways involved with both UBE3A activity and structural brain integrity.
Protein analysis identified UBE3A as the sole significantly downregulated protein present within the Ube3amQ606E cortex, marking it unusually distinct compared to control mice. This downregulation, combined with the hearty upregulation of proteins such as HERC2—typically associated with neurodevelopmental alterations—indicates how Ube3amQ606E significantly influences the brain's molecular milieu at large, potentially fostering deleterious effects over time.
This study signifies historically unexplored impacts of UBE3A gain-of-function on neurodevelopment, drawing new connections with Angelman syndrome's pathology. With UBE3A's effects ubiquitous and influential, therapeutic exploration must shift to account for both loss-of-function and gain-of-function variants—especially considering potential pathways aimed at reinstatement of UBE3A function for patients exhibiting these neurodevelopmental differences. The findings pave the path forward for innovative therapeutics targeting genetic expressions of UBE3A.
Future inquiries are warranted to challenge existing knowledge surrounding UBE3A's functional dynamics, wherein therapeutic strategies could leverage insights from both genomic mutation profiles and broad molecular effects on cognition and behavior as observed.