Recent research has unveiled that a deficiency in the enzyme HSD17B4 impairs the formation of primary cilia, which are crucial cellular structures involved in sensing and signaling. This inefficiency in ciliogenesis hints at intricate connections between cellular metabolism and the development of ciliopathy-like symptoms, such as impaired motor function and developmental delays. The study highlights the role of acetyl-CoA, generated through peroxisomal β-oxidation, in restoring ciliary function and potentially serving as a therapeutic target in related genetic disorders.
Primary cilia are essential organelles found on the cell surface, playing instrumental roles in various physiological processes. These organelles mediate key signaling pathways relevant not only to cell growth and differentiation but also to the overall health of tissues and organs. When primary ciliogenesis is disrupted, a slew of genetic disorders known as ciliopathies arises, characterized by symptoms such as kidney disease, retinal degeneration, and developmental anomalies, including motor coordination deficits.
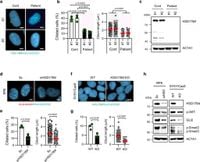
The enzyme HSD17B4 is vital for peroxisomal metabolism, specifically in the process of breaking down fatty acids and generating metabolic intermediates essential for energy. Deficiencies in this enzyme lead to significant metabolic dysfunction and a spectrum of symptoms similar to those resulting from ciliopathies. According to the recent publication in Nature Communications, researchers have linked HSD17B4 deficiency with impaired ciliogenesis, suggesting a direct functional relationship between ciliary health and peroxisomal metabolism.
In-depth experiments revealed that primary fibroblast cells derived from patients with HSD17B4 deficiency exhibited drastically reduced ciliation. Such findings were mirrored in both human-derived retinal pigment epithelium (RPE) cells and engineered SH-SY5Y neuronal cells lacking HSD17B4. Quantitative analysis showed fewer cilia and diminished ciliary length in defective cells, reinforcing the impact of HSD17B4 on ciliogenesis.
The study provides powerful evidence that disruption of peroxisomal β-oxidation significantly impairs the formation of primary cilia. Additionally, factors affecting cilia development—like HDAC6-mediated deacetylation of tubulin—were also examined. Researchers found that the restoration of acetyl-CoA levels, either through direct supplementation or metabolic manipulation, could mitigate ciliary defects. Acetate treatment, particularly, effectively increased primary cilia formation and length in HSD17B4-deficient models.
Captivatingly, vivo studies in mice demonstrated the therapeutic potential of acetate administration. Hsd17B4-knockout mice, which display severe developmental defects and high mortality, exhibited improved motor function and enhanced ciliary formation when treated with acetate during critical developmental stages. Survival rates doubled among treated mice, underscoring the influence of metabolic health on ciliopathy-like symptoms.
Cumulatively, these findings shine a spotlight on the pivotal connection between peroxisomal metabolism, primary ciliogenesis, and cellular health. As the research delineates, therapies that elevate acetyl-CoA or modulate pathways involved in ciliary dysfunction could represent promising avenues for addressing genetic conditions linked to HSD17B4 deficiency. Future studies are needed to elucidate the precise mechanisms and potential clinical applications of acetate as a metabolic supplement in the treatment of ciliopathy-related disorders.