An Innovative Cellular Assay Shows How Specific MFN2 Variants Affect Mitochondrial Function in Charcot-Marie-Tooth Disease
New findings offer insights into the genetic underpinnings of a common hereditary neuropathy, potentially improving patient diagnosis and treatment.
Charcot-Marie-Tooth disease (CMT) is among the most frequently diagnosed hereditary neuropathies. Now, researchers have made significant strides in understanding the genetic factors involved, particularly with a subtype known as CMT type 2A (CMT2A), by developing a cellular assay to evaluate the effects of various mutations in the MFN2 gene.
CMT2A, a hereditary disorder primarily affecting peripheral nerves, has been linked to mutations in the MFN2 gene, which encodes a protein crucial for mitochondrial fusion. Using next-generation sequencing, an increasing number of MFN2 variants have been identified, but many remain classified as variants of uncertain significance (VUS), leaving patients without a clear diagnosis.
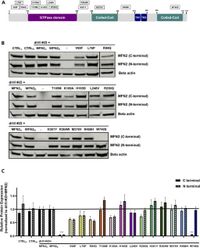
In a study published on March 22, 2025, in Scientific Reports, researchers led by Barsa, Perrin, and David assessed 12 MFN2 variants previously identified in patients with CMT2A. Their novel assay employs double mutant knock-out mouse embryonic fibroblasts (dMfnKO MEFs) to measure the mitochondrial fusion capacity of these mutations. Surprisingly, only six out of the twelve tested variants displayed a reduced ability to facilitate mitochondrial fusion, suggesting diverse impacts of these mutations on cellular function.
The analysis revealed that variants classified as ‘fusion-defective’ were often associated with an earlier age of onset for CMT2A, while ‘fusion-competent’ variants correlated with later onset. “The classification of variants as ‘fusion-defective’ or ‘fusion-competent’ appeared to correlate with the age of onset observed in CMT2A patients carrying these SNVs,” wrote the authors of the article. This correlation provides potential pathways for further understanding the clinical diversity in patients diagnosed with CMT2A.
In particular, researchers focused on variants such as p.R94Q, p.T105M, and p.H361Y, which had known pathogenic effects in other studies. These mutations stand out due to their complete loss of fusion capacity, which aligns with a severe clinical presentation of CMT2A. Conversely, mutations like p.R468H and p.R250Q displayed sufficient mitochondrial fusion activity, suggesting they are less likely to be pathogenic.
As the field of genetics rapidly evolves, the capability to distinguish between pathogenic and benign mutations is critical for patient outcomes. The researchers utilized functional characterization methods that enabled reliable comparisons among different MFN2 gene variants. Their findings articulate a critical step towards refining genetic counseling and precision medicine approaches for affected families.
Furthermore, the computational tools used to predict the impact of these mutations were evaluated in terms of their effectiveness. The study found that predictions aligning with functional assay results generally provided a more accurate classification of pathogenicity. “Our future goal will be to improve the sensitivity and resolution of the cell-based functional assay and to widen the scope of this test by investigating further MFN2 variants,” wrote the authors of the article, suggesting an ongoing effort to enhance understanding of genetic variances in CMT2A.
As CMT remains a prevalent hereditary condition affecting numerous individuals globally, the urgent need for effective diagnostic tools is paramount. This study not only advances current knowledge but paves the way for future research, focusing on the complexities of mitochondrial dynamics and genetic factors influencing neurological health.