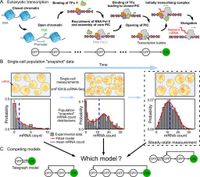
Mathematical modeling has provided a nuanced understanding of transcription mechanisms within eukaryotic cells, but a recent study emphasizes the limitations of using steady-state mRNA count data to estimate the number of inactive gene states. Such estimations are critical for grasping how genes are regulated during expression, yet, as the findings suggest, they may not be as straightforward as previously thought.
The essence of transcription in eukaryotic cells involves a complex series of events that lead to the production of mRNA from DNA, which then plays a vital role in protein synthesis. Despite advancements in revealing these processes, significant challenges remain in accurately quantifying the number of inactive states a gene may occupy prior to becoming active.
The research, which draws upon a suite of mathematical models tailored to fit data from mRNA counts, found that most of the steady-state distributions could indeed be replicated with a model featuring just a single inactive state. This is particularly problematic for scientists seeking to understand the multifaceted nature of gene regulation, as it obscures insights into the actual mechanisms at play.
"It is currently unclear if by fitting steady-state distributions only, the number of rate-limiting steps and the network topology of the true transcriptional mechanism can be identified," the authors of the study noted, capturing a critical limitation of existing methodologies.
The research team conducted a series of simulations, dissecting the parameters that govern transitions between active and inactive states in genes. They demonstrated that for many eukaryotic cells, the initial response to transcriptional induction may follow a power law, where the slope of the relationship between mean mRNA count and time is indicative of the gene states navigated during this transient phase.
This approach could simplify the inference of gene states by relying less on extensive temporal data and allowing for the characterization of gene expression dynamics over shorter intervals, which are often more manageable in experimental settings. By focusing on the initial rise and slope of mRNA count after induction, the researchers unveiled a new pathway to estimate the potential complexity of gene regulation.
Moreover, these insights have the potential to transcend the specific cases studied, as the model framework might also be applicable to various biological systems beyond the scope of the original research, enhancing our overall understanding of gene expression in other organisms.
As the research community looks to elaborate upon these findings, it underscores an important reality: while mathematical models can elucidate biological phenomena, they must be applied judiciously. The work sets a strong foundation for future inquiries into transcription mechanisms, and opens the door to refining approaches that could yield richer insights into how genes are regulated in the intricate tapestry of cellular activities.
In conclusion, the limitations of traditional methods to estimate inactive gene states using steady-state mRNA counts lead to exciting new avenues for understanding the collective dynamics of gene regulation, with profound implications for genetics and functional biology.